



















Blogs
Somatic Variant Classification: Mining for Clues
Taking a closer look at somatic mutations, we see how variant databases can serve as rich resources for variant-related information. The challenge lies in using these data to determine whether a specific variant is harmful, warrants attention, or is mostly harmless. This leads us directly to the issue of variant classification.
Next-Generation Sequencing (NGS) protocols analyze huge volumes of DNA and identify deviations from a reference genome, ranging from single nucleotide variations to copy number changes. Genomic sequencing throws up thousands of such variants, but only a handful of them are clinically relevant. Variant classification is the systematic process by which clinically significant, potentially oncogenic variants are winnowed out from a patient's genetic data to make treatment decisions. Variants can be classified into five categories: oncogenic, likely oncogenic, variant of unknown significance, likely benign, and benign.
Given the crucial nature of variant classification, experts from around the world have collaborated to establish standard rules for classifying variants, such as the AMP, ASCO, and CAP joint consensus guidelines. These guidelines specify the parameters that determine whether a variant is harmful and define the 'level of evidence' required [1].
Consider, for instance, the functional effects of a gene mutation—a crucial parameter in variant classification. Functional data, gathered from reported experiments in medical and epidemiological literature and predicted by computational algorithms like FATHMM and Mutation Taster, can indicate how mutations might alter protein structure and function. The greater the evidence of functional alteration, the higher the likelihood of the mutation being oncogenic. Other key parameters include population frequency and the presence of cancer hotspots [1].
Interpreting a patient’s variant data into actionable knowledge requires expert teams to deliberate numerous parameters, utilizing databases with millions of constantly updated data points. This process is expensive and time-consuming, and any delay in initiating therapy could worsen a patient's prognosis.
An automated process that systematically goes through these steps to yield quick and accurate results could be a true game changer. The StrandIris platform is a nearly automated HIPAA-compliant tool that can sift through a patient’s NGS data to report a comprehensive profile of actionable variants and therapy recommendations, with a remarkably short turnover time of a few minutes.
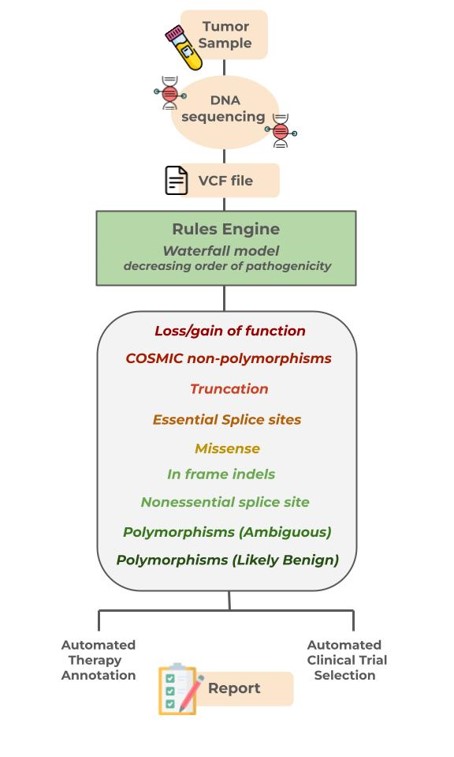
The Iris platform integrates a rules engine with over 200 intricate algorithms for classifying variants, to a knowledge base harboring more than 24,000 variants and 2300 therapy recommendations. Sequence data passes through a waterfall model of ten categories in decreasing order of pathogenicity, including loss/gain of function, essential splice site changes, missense mutations, and more. Each category must meet all its conditions before moving to the next. Unlike other platforms, Iris can factor in polypeptide length, sequence, stop codon location, and protein domain architecture to determine if a variant could result in upregulation, downregulation, or loss of function.
After classifying variants using these rules, the platform pairs each variant with recommended drugs or treatments. Tumors often progress to become genetically heterogeneous, harboring numerous mutations. These mutations can interact, influencing how resistant or sensitive a patient might be to a particular treatment course. StrandIris adeptly taps into this complexity, basing its final therapeutic recommendations on the comprehensive mutational profile of the patient. This capability provides clinicians with invaluable tools for personalized medicine—and in the next segment, we shall find out why!
You can find more details regarding StrandIris, here!
Explore the rest of the blog series below:
- Part 1: Precision Oncology: A Genetics Revolution in Cancer Management
- Part 2: Somatic Variants and Databases: A Wealth of Information
- Part 4: Somatic Variants: Towards Better Therapy
References:
1. Horak, P., Griffith, M., Danos, A. M., Pitel, B. A., Madhavan, S., Liu, X., & Sonkin, D. (2022). Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genetics in Medicine, 24(5), 986-998.

Precision Medicine
30 Jun 2025
Strand’s Bioinformatics Expertise Enables Liquid Biopsy Assay Development for NSCLC with 70% Cost Reduction and 2–5% VAF Sensitivity
Precision Medicine
08 Apr 2025
How Genetic Analysis Is Shaping the Future of Xenotransplantation
Precision Medicine
12 May 2026
Biologicals and Microbials in Agriculture: Understanding the Science and the Data Behind Them
Let's Connect
Let's Connect

download the case study.