



















Blogs
AI or Manual Curation: The Path Forward
Our genetic differences shape our identity and health, influencing traits, disease risks, and treatment responses, making it essential to identify relevant variations for personalized healthcare. From detecting rare genetic diseases, to assessing cancer or cardiovascular risk, to predicting the body’s response to a range of drugs, variant detection and classification lies at the heart of this revolutionary era in precision diagnostics and medicine. Classification refers to the process of categorizing genetic variations based on their potential impact on health and disease, and is done following the standardized guidelines set by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP).
Yet, the interpretation of genetic screening tests is not as smooth and painless as it would seem. The speed, efficiency and cost effectiveness of NGS has resulted in a colossal expansion of detectable genetic variants, and experimental techniques such as deep mutational scans or multiplexed assays of variant effects (MAVEs) have parallelly thrown up data about their functional effects. Each year, thousands of biomedical publications exploring gene-disease links are added to pubmed. There are multiple databases that aim to house this information such as Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database (HGMD), ClinVar, etc - each of which have their own criteria for variant inclusion, and ontologies for variant description. Interpreting the clinical significance of genetic variants by collating information from multiple, dense data sources is the most time-consuming and costly part of genetic diagnosis.
Given its superhuman capacity to process big data, can AI be the panacea we need - potentially removing the need for manual attention at all while curating variants?
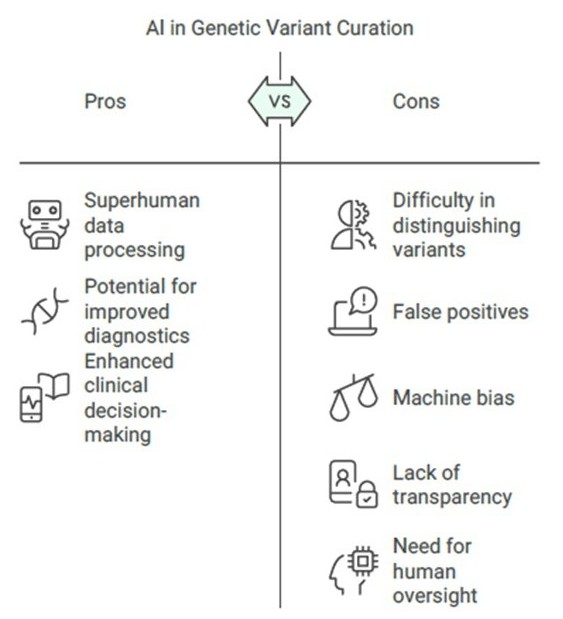
Artificial Intelligence, especially deep learning algorithms, have helped us rise up to the challenge of large, complex datasets. AI encompasses simpler machine learning algorithms such as regression, as well as deep learning which uses neural networks. These algorithms, which can directly take as its input DNA sequences, are trained on huge datasets with little requirement for humans to extract or engineer predictive features for it. Convolutional Neural Network models have been used to aid document triage and variant evidence extraction from literature, and automated curation or text-mining processes often yield a much higher number of unique variants than manually curated databases. Deep learning, computer vision techniques and natural language processing can help analyze genomics-related information found in scientific publications/data across sweeping volumes and in exponentially less time.
Given its superhuman capacity to process big data, can AI be the panacea we need - potentially removing the need for manual attention at all while curating variants? While it is an exciting thought, taking a closer look would reveal that for this, there is some way to go.
The main goal of genetic variant databases in healthcare is to help diagnose diseases and predict future disease risks. This means we need to consider the quality of the data, and its ability to allow meaningful and relevant genotype-phenotype connections without inaccuracies and biases. This is where things become challenging with an entirely AI based setup. AI tools often struggle with distinguishing between clinically relevant variants and those that are benign, as they rely on existing data and algorithms that may not capture all nuances of genetic variations. They have been known to throw up numerous false positives resulting from incomplete gene annotations, or variation either in gene nomenclature or the transcript being considered, leading to inaccuracies due to shifted positions. These differences have to be spotted and manually resolved by referring to literature, supplementary material, variant databases etc. Many clinically irrelevant variants also get added to the pool, increasing volume without improving practical outcomes. Advanced AI algorithms are sometimes not transparent, and therefore assessing their accuracy, context relevance, and generalizability becomes tricky.
Another area of concern is machine bias. Genomic and health data often contain substructure influenced by both true causal relationships and external factors like socioeconomic status or unequal access to healthcare. AI systems must be carefully managed to avoid biases from these factors, as unequal representation in training data can lead to biased genetic risk predictions and worsen disparities in healthcare.
More informatics research and development is required to fully exploit the therapeutic promise of AI technologies. In order to replicate AI's achievement in other domains without exacerbating racial, ethnic, or socioeconomic biases, the emphasis should be on utilizing these technologies in conjunction with physicians to enhance clinical decision-making, considering the intricacy and unpredictability of genetics and medicine. The most optimum approach seems to be a hybrid one, where the output of AI based tools are processed and assessed manually by variant scientists.
At Strand, putting together the best of both worlds is our forte. Our genomic testing and variant classification system utilizes top of the line computational tools together with a team of expert variant scientists who figure out the actual clinical relevance of the genetic variants in an individual.
Strand's Somatic Oncology Knowledgebase provides detailed, regularly updated information on over 16,000 gene variants, drugs, and clinical trials, enabling faster, data-driven clinical decisions. It integrates FDA-approved and investigational drug recommendations, including clinical trial details, and supports identifying effective treatments for patients with variant combinations or drug resistance. The platform offers automated annotation of novel variants based on functional significance and hotspot positions, and includes evidence-based annotations, periodic updates, and customizable reporting tools such as StrandIris, a nearly automated HIPAA-compliant solution for the reporting of variants from oncology NGS panels. StrandIris incorporates the position of the variant in relation to the protein domain architecture and ensures that the functional significance of the variant is elucidated to the highest extent possible. Thus, even variants with limited published evidence are categorized based on their functional and/or pathogenic impact. Our team of variant scientists use these tools and their knowhow to ensure accurate information about precision genomics reaches our clients, as quickly as possible.



Let's Connect
Let's Connect

download the case study.