




















Blogs
FDA Final Rule Affects Regulatory Oversight of LDT Manufacturers
Quick Overview:
- The FDA announced a final rule that classifies LDTs as IVDs. Thus, LDT manufacturers now need to comply with more regulations.
- The new regulations are a result of the FDA's intention to improve patient safety.
- A four-year plan to phase out the previous enforcement discretion policy was stated.
On April 29, 2024, the U.S. Food and Drug Administration (FDA) recently announced its final rule affecting the oversight of CLIA (Clinical Laboratory Improvement Amendments) laboratory-developed tests (LDTs).
What are Lab Developed Tests?
LDTs are a type of in vitro diagnostic (IVD) tests that are completely designed, manufactured, and used within a single CLIA-certified laboratory. They are not sold or distributed to any other laboratories or health care facilities, unlike commercial laboratory tests.
To elaborate, they are assays conducted on biological samples for the purpose of diagnosis and informing clinical decisions.
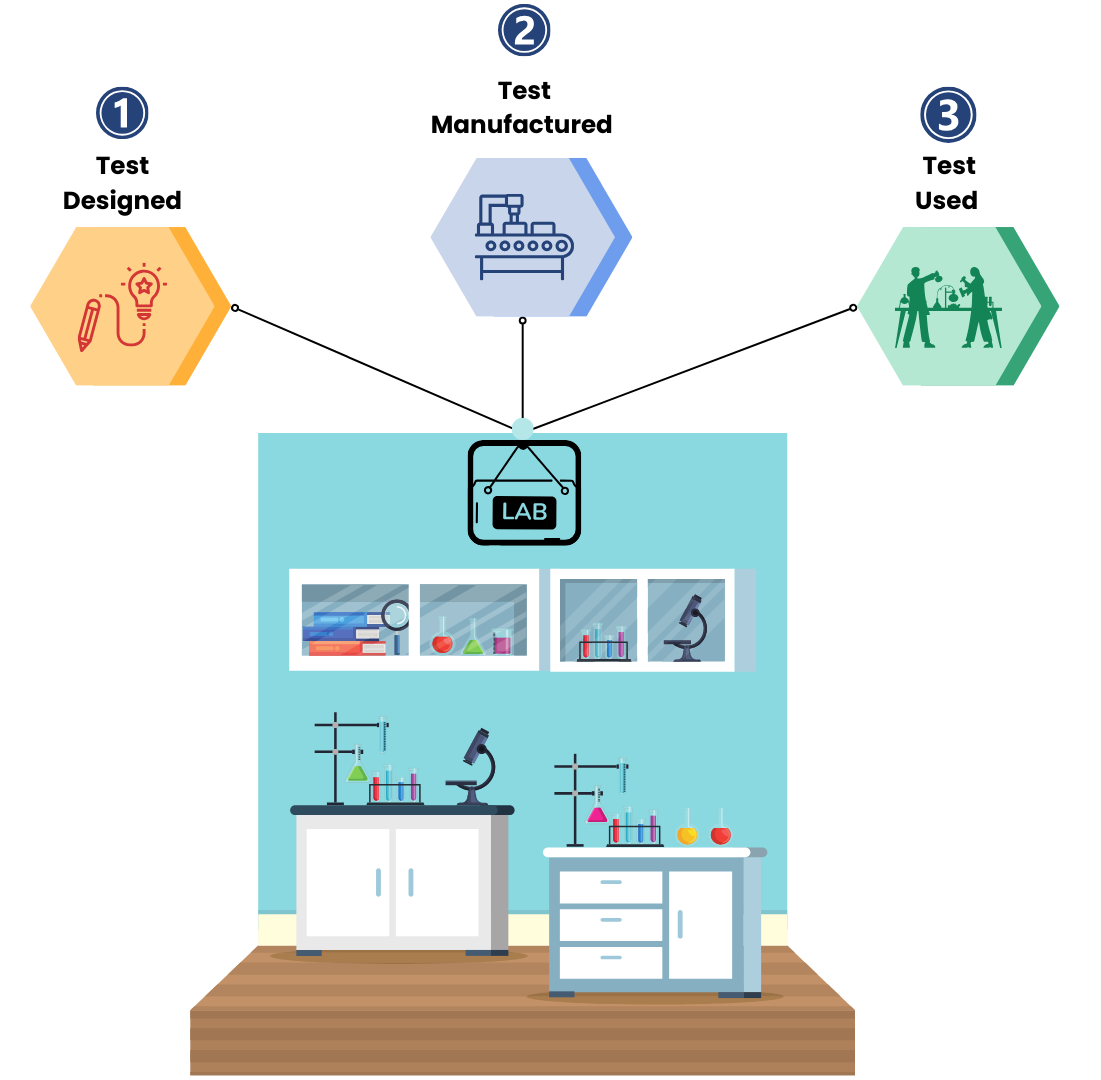
LDTs are laboratory tests developed and used within the same lab that creates them
What does the Final Rule State?
The new ruling extends the definition of IVDs and explicitly states that LDTs are considered IVDs under the Federal Food, Drug, and Cosmetic Act.
Thus, the FDA now requires LDTs to comply with FDA regulations for IVDs.
What do these regulatory changes mean?
Before the publication of the final rule, the FDA had exercised an enforcement discretion policy for LDTs. The concept of enforcement discretion means that the agency had chosen not to enforce its regulations for LDTs, even though the regulatory requirements do apply.
However, in April 2024, the FDA announced a phase-out of the enforcement discretion, citing concerns about the reliability and efficacy of these tests. With the final rule, the FDA intends to improve healthcare decisions and ensure patient safety.
How does the FDA plan to phase out enforcement discretion?
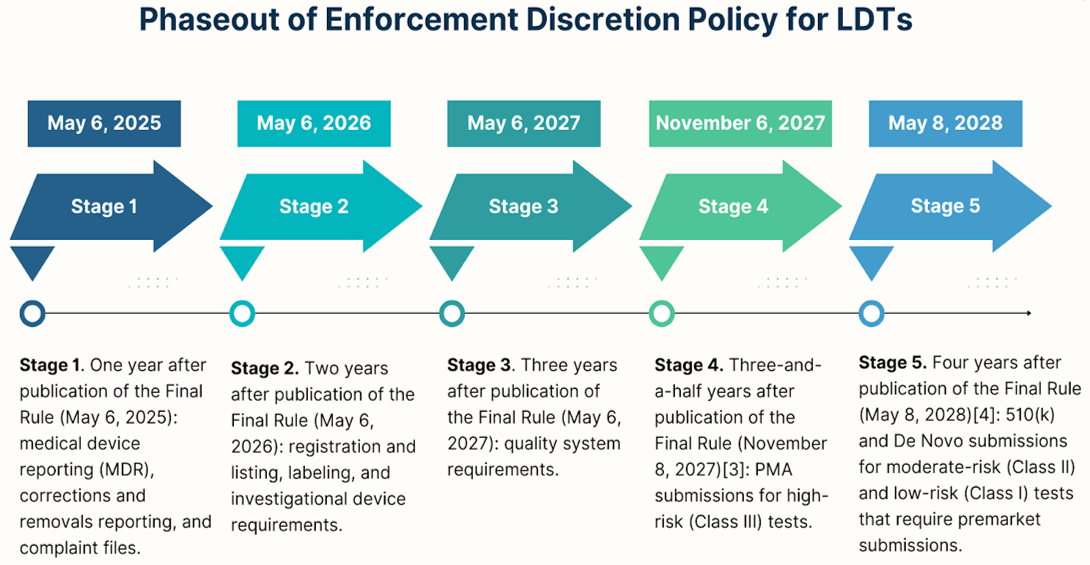
The FDA's final rule establishes a four-year, five-stage plan for phasing out enforcement discretion for LDTs. Each stage of the phase-out policy enforces a successively greater degree of compliance over four years from the rule's publication on May 6, 2024.
Here are the key takeaways:
- Stage 1: By the end of one year (May 6, 2025), the FDA will require laboratories to comply with Medical Device Reporting (MDR) requirements, correction and removal requirements, and Quality System (QS) requirements regarding complaint files.
- Stage 2: Two years following the final rule (May 6, 2026), the FDA will expect laboratories to abide by registration and listing requirements, labeling requirements, and investigational use requirements.
- Stage 3: By the end of three years (May 6, 2027), the FDA will end enforcement discretion for QS requirements other than those for complaint files.
- Stage 4: After three and a half years since the final rule (November 6, 2027), laboratories are expected to meet premarket requirements for high-risk (Class III) IVDs offered as LDTs or receipt of a premarket submission to the FDA prior to the start date, in which case enforcement discretion applies pending FDA review of the submission.
- Stage 5: Four years after the final rule (May 8, 2028), the FDA will end enforcement discretion regarding premarket review requirements for moderate-risk and low-risk IVDs offered as LDTs that require premarket submissions, or receipt of a premarket submission prior to the beginning of this stage, in which case enforcement discretion applies pending FDA review of the submission.
Do these regulations apply to all LDTs?
While it is important for LDT manufacturers to prepare and orient themselves in this new regulatory landscape, it is also worth noting that these rules do not apply to all LDTs.
The FDA will continue to exercise enforcement discretion for certain kinds of LDTs. In our next blog, we will delve deeper into the different types of LDTs and the corresponding regulatory expectations.
Precision Medicine
15 Jan 2025
Strand's festiVAR Tool Achieves 40% Diagnostic Yield in Comprehensive Neurological Exome Sequencing

Precision Medicine
23 Jan 2024
Going from Bench to PDF with Strand's PQRS software

Precision Medicine
06 Dec 2024
Strand’s Automated Variant Verification System Cuts Down Efforts by 80%
Let's Connect
Let's Connect

download the case study.