




















Blogs
Partnering with New Molecular LDTs: Strand's FDA Compliance Roadmap
In the previous two blogs, we discussed the FDA’s 4-year, 5-stage phaseout of enforcement discretion policy and which LDTs need to abide by the various rules.
In this final blog, we will discuss the FDA’s rules for new molecular LDTs in more detail, the processes that need to be established, and lastly, how Strand can help.
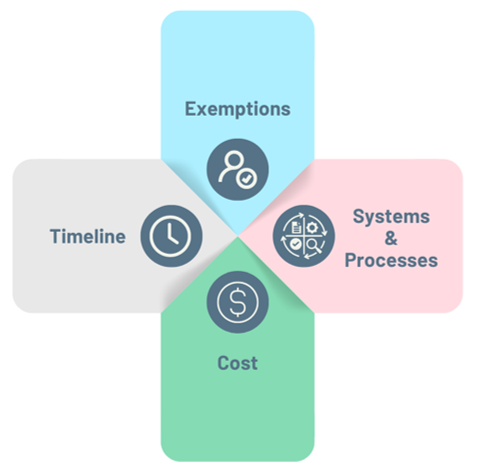
If you are a new laboratory seeking to develop, manufacture, and market LDTs after the publication of the Final Rule, these are the various puzzle pieces that need to be resolved:
- Exemptions: The LDT manufacturer will need to determine to what degree of exemption, if any, they qualify for.
- Systems and Processes: Based on the answer 1), they will need to set up reliable processes to accompany the LDT and comply with the final rule.
- Timeline: Based on the above, the stages of enforcement discretion phaseout will need to be put into place.
- Cost: Based on the answers to b and c, the costs of setting up these processes will have to be estimated.
Below, we will elaborate on these 4 factors:
Exemptions
1. Exempt from all requirements
a. Your molecular test is exempt from all final requirements if it is a HLA test.
b. Your molecular test is a Tier 1 test as covered in Part 2 of this series.
2. Exempt from premarket review requirements
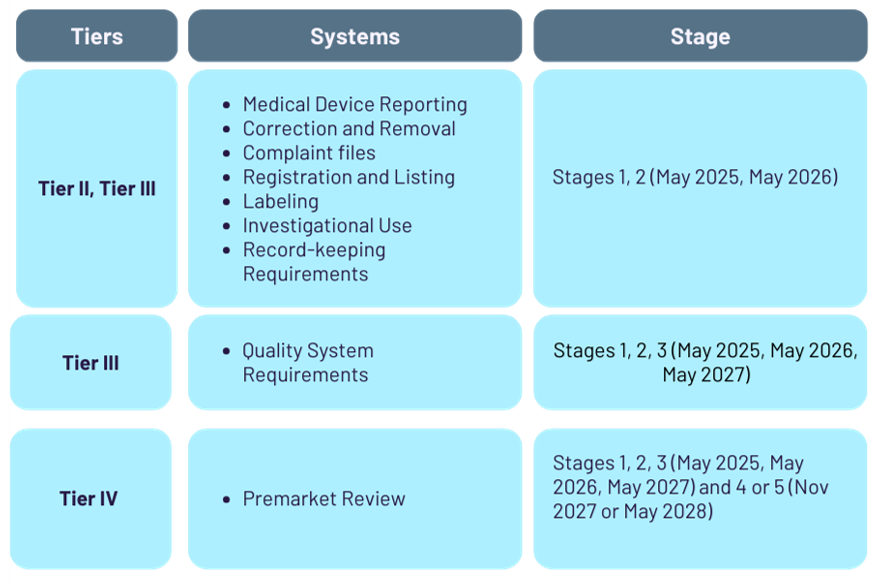
Your test is exempt from premarket review requirements if it's either a Tier 2 or 3 test. For new molecular tests, this generally implies two further possibilities:
(i) *LDTs for unmet needs manufactured and performed by labs integrated in the healthcare system treating the patient. This requires fulfilling two criteria:
- Does my LDT fulfill an unmet need?
- Will my test or can my test be integrated into the healthcare system treating the patient, i.e., a hospital or an academic medical center (AMC)?
(ii) LDTs that have NY state CLEP approval.

If your lab is in the state of New York or is otherwise seeking to market its LDT in the state of New York, it will need NY State CLEP approval. Obtaining NY State CLEP approval will further exempt your LDT from the onerous premarket approval needed in either Stage 4 for high-risk or Stage 5 for medium and low-risk LDTs.
*These are further exempt from most quality system requirements needed by Stage 3, except record-keeping and complaint file-keeping requirements.
Systems and Processes & Timeline
Most new molecular LDT manufacturers will not be exempt and hence fall under one of the Tiers II, III, or IV.
Cost
After a new molecular LDT lab has clarified the required systems and processes that need to be set up and honed in on the timelines, it is valuable to estimate the cost. The total expenditure for compliance to the new rule would depend on various factors:
- A detailed description of the LDT
- Accompanying descriptions/documents of validation of LDT, CLIA reports, etc
- Planned LDTs
- Laboratory systems currently in place
- Compliance to MDR, correction and removal, QS, labeling, Investigational Use, etc currently in effect
- Laboratory budgets by year for new systems
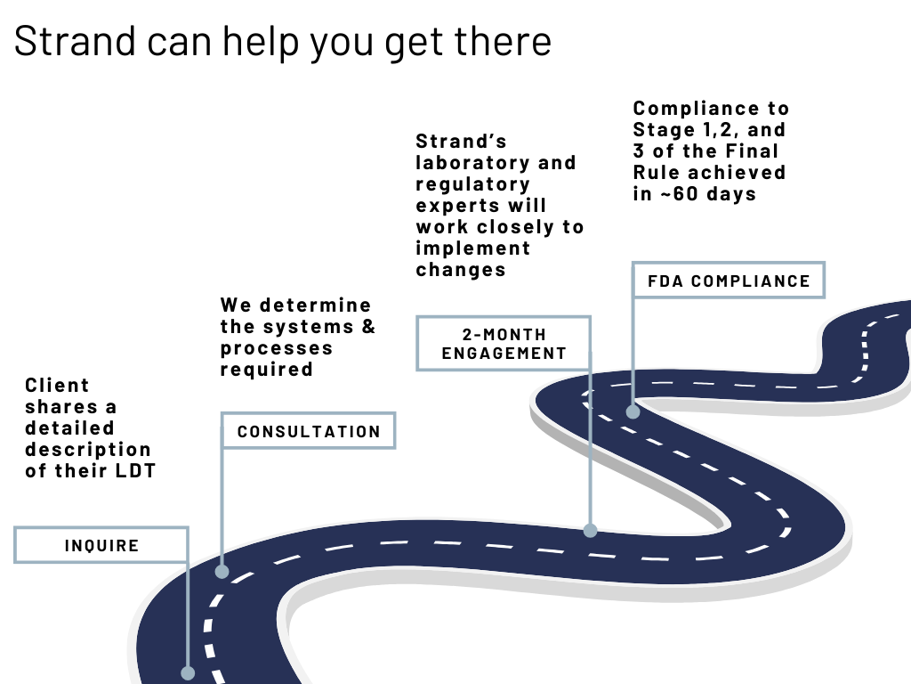
Strand has developed (extensive knowledge of) FDA regulations and experience in establishing LDTs.
Depending on the type of LDT and the description provided, Strand will consult with you in a 2-month timeframe to determine the systems that need to be put in place for a phased compliance with Stages 1,2, and 3 of the Final Rule. This 2-month exercise will comprise Strand’s laboratory and regulatory consultation.
Additionally, if the test requires either Stage 4 or 5 premarket review compliance, Strand will interface with an FDA consultant to come up with a plan for such compliance, as well as timelines.
The initial engagement will cover Stage 1-3 consultation and stage 4 and 5 compliance at a later phase, as the latter is not needed before Nov 2027.
Explore the rest of the blog series below:


Let's Connect
Let's Connect

download the case study.