



















Blogs
Spatial Proteomics
End of year celebrations bring in reflections of the year past and excitement for the upcoming year. In the world of biotech this is exemplified by Nature’s choice of method of the Year: Spatial Proteomics. While spatial technology has been around for a few years now, spatial proteomics looks specifically at how protein structures exist and interact in 3D spaces in various contexts such as cells, tissues and organ slices. It actually encompasses a large group of both older and newer technologies including co-detection by indexing (CODEX), iterative bleaching extends multiplexity (IBEX), imaging mass cytometry (IMC), multiplexed ion beam imaging (MIBI), cyclic immunofluorescence (cycIF), and the more recent deep visual proteomics (DVP); making it the perfect candidate for something that builds on the past and promises exciting avenues for the future.
We recently developed a custom R-based workflow tailored for analyzing spatial transcriptomics data.
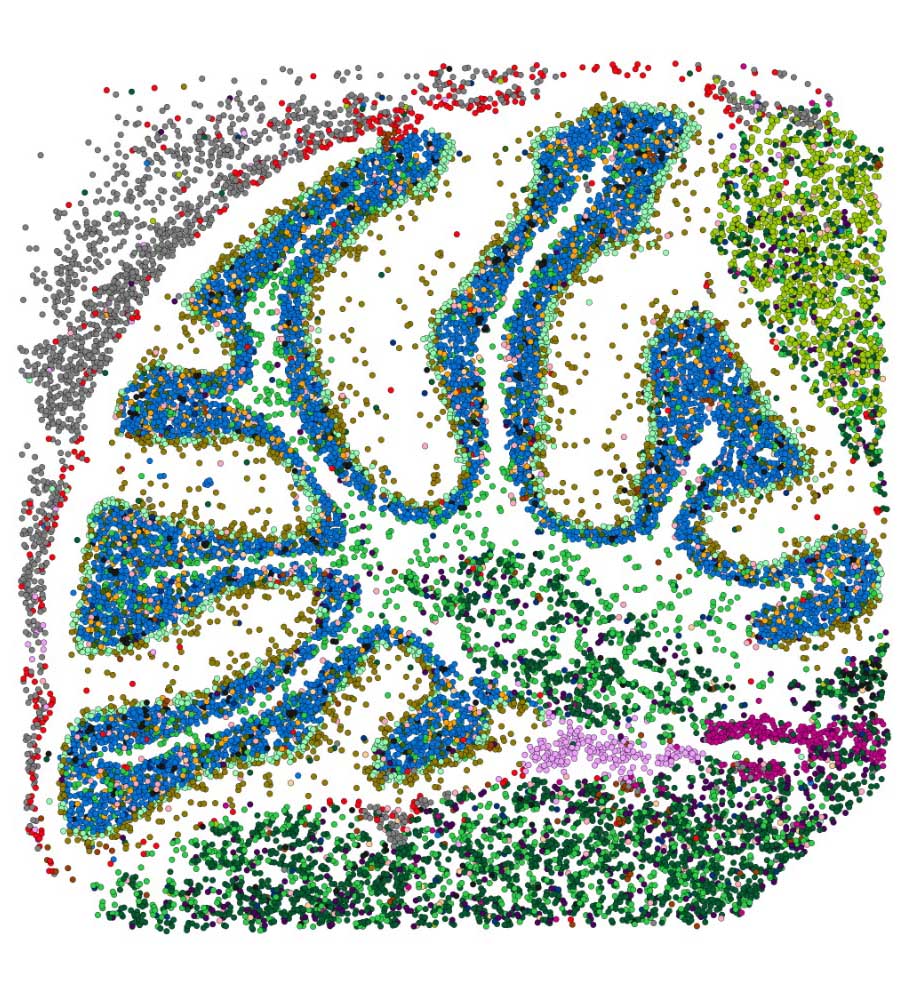
Here at Strand, we have been working in the spatial domain for a few years now, including our spatial transcriptomics pipeline. Spatial transcriptomics (ST) is an advanced technique that integrates microscopy and sequencing to map gene expression while maintaining the spatial context within tissue samples. This approach offers valuable insights into biological processes in both healthy and diseased states, insights that traditional methods cannot provide.
We recently developed a custom R-based workflow tailored for analyzing spatial transcriptomics data. The comprehensive computational pipeline covers all stages of data analysis, including raw data processing, quality control, normalization, batch effect correction, identification of spatially variable genes, and cell type annotation, providing a thorough end-to-end solution.
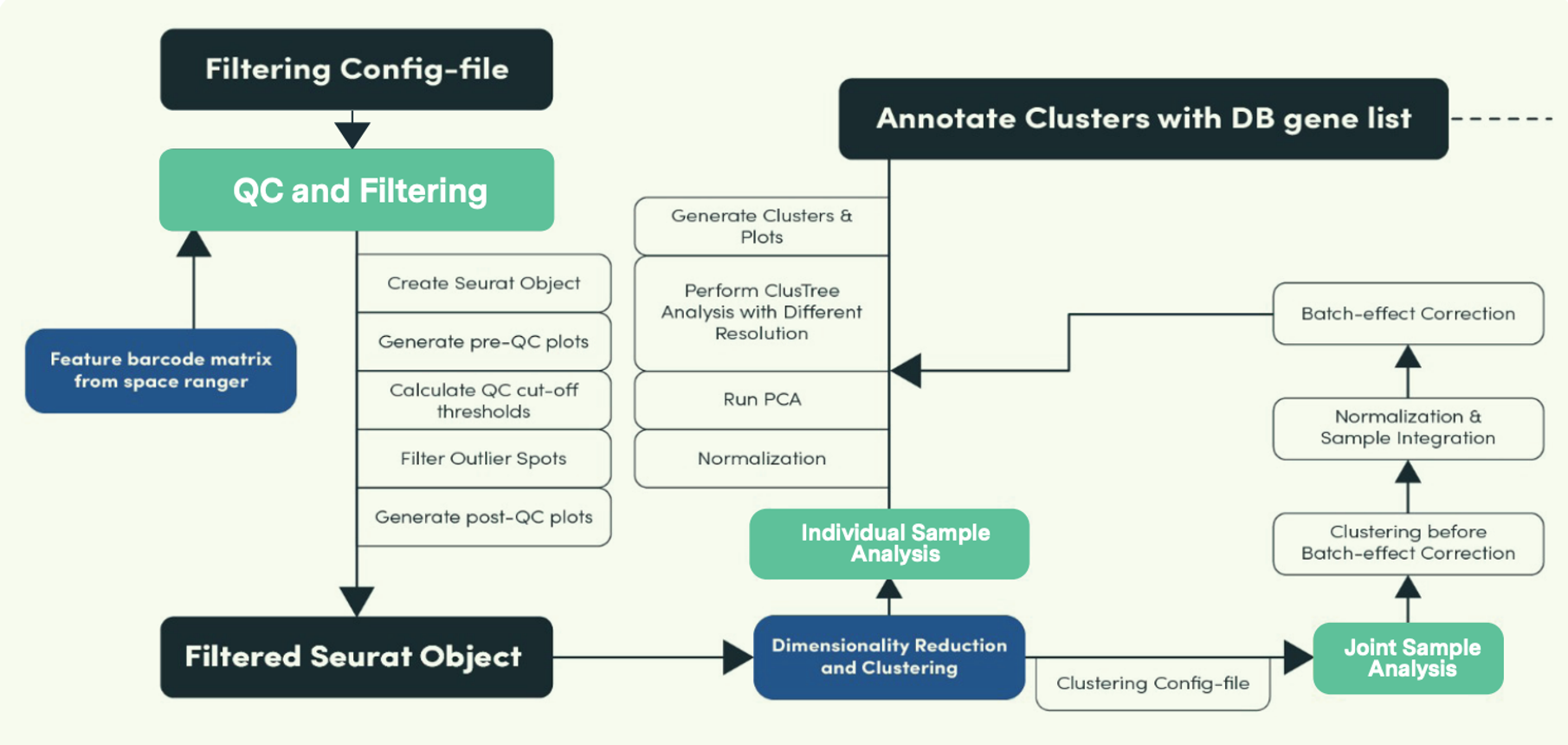
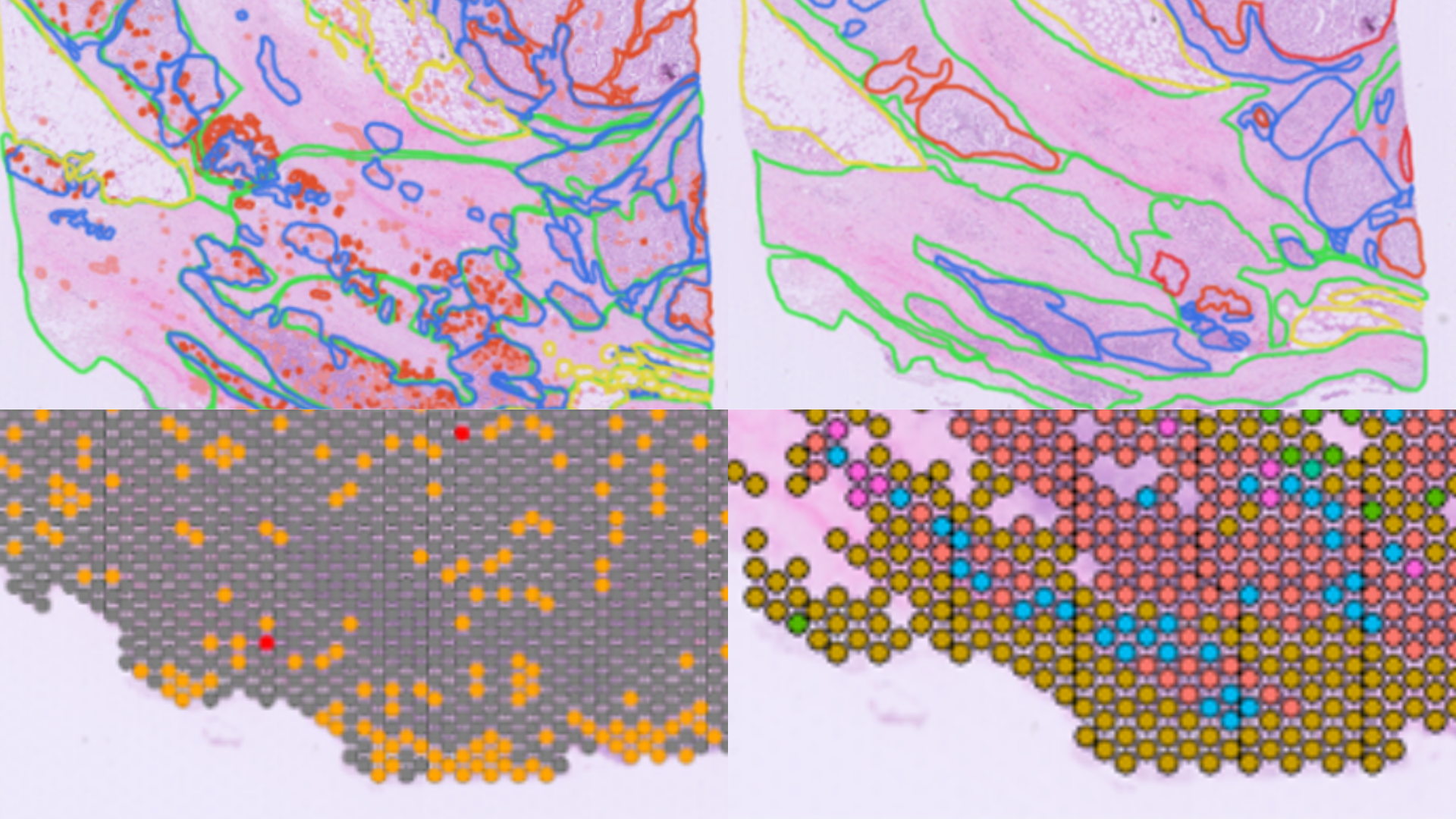
As illustrated in the image above, this pipeline has three main modules (highlighted in green), QC and Filtering, Individual Sample Analysis and Joint (or combined) Sample Analysis. To tackle typical challenges seen in spatial transcriptomics such as identifying cell types and accurately locating them on tissue slides, the pipeline incorporates various annotation algorithms, including ScType, SingleR, Seurat label transfer, and robust cell-type decomposition (RCTD). Check out our recent webinar detailing how it works here.

Additionally, our pathology, molecular biology, and bioinformatics teams successfully validated the 10X Genomics Visium workflow in FFPE samples; wherein we assessed the correlation between spatial gene expression and immunohistochemistry, as well as histopathological annotations. This workflow led us to become a Certified Service Provider for 10X Genomics. You can learn more about it in this case study.
We’re excited to see what the new year brings, and the advancements that will be made in this exciting field!


Let's Connect
Let's Connect

download the case study.