



















Blogs
Strand’s Methylation Pipeline - An Overview - Part 1
We developed a methylation pipeline on AWS Healthomics. The first part of this two-part series outlines the steps in the pipeline for analyzing methylation data from targeted methylome sequencing (TMS) and whole methylome sequencing (WMS), and the second part walks through the protocol for users to run this workflow on AWS Healthomics.
Why did we choose AWS HealthOmics?
We chose to leverage AWS Healthomics to optimize storage and computing costs. Storage on AWS is 75% lower than S3, and compute is 25% lower than EC2. The table below summarizes the additional benefits:

Summary of the pipeline
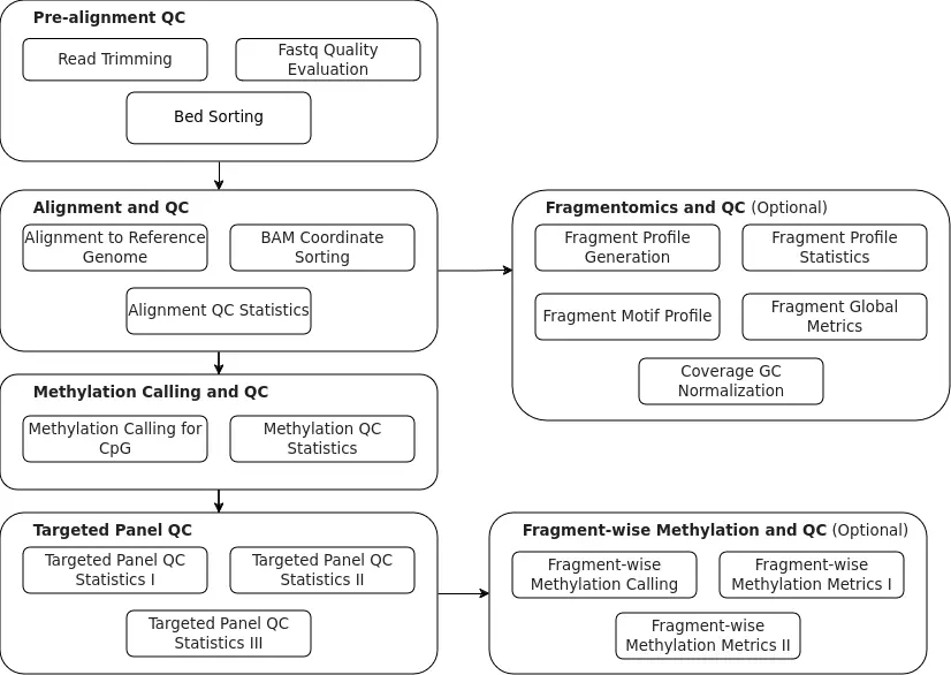
Our pipeline consists of four main analysis phases, along with two optional steps:
1. Pre-alignment QC
-
- Here, the processes of trimming raw reads and FASTQ quality evaluation are carried out. BBDuK trims the FASTQ files based on quality and the presence of adapter sequences, and FastQC generates a quality control (QC) report from the FASTQ files to assess sequencing quality.
2. Alignment and associated QC
- Essential alignment of the FASTQ files to the reference genome is done using BWAMeth.
Fragmentomics (optional): Alternatively, depending on the user’s research questions, the aligned file from stage 2 can be used for fragmentomics analysis and QC. FranaTK generates a range of fragmentomics features.
3. Methylation calling for CpGs
-
- Methylation calling for CPGs is handled by Methyldackel, which generates CpG, CHH, CH, and bedgraph files.
4. Targeted panel analysis
- The final step includes targeted panel analysis and producing related QC metrics.
Fragmentomics (optional): An option to proceed with fragment-wise methylation calling is available after stage 4. The tool Patr generates fragment-wise methylation patterns and summaries.
The above pipeline is summarized in the following figure:
Tools in the pipeline:
- BBDuK: Remove contaminating adapter and trim low quality regions
- FASTQC: Generate QC distributions
- BWAMeth: Read Alignment
- Picard: Generates alignment and enrichment metrics
- FranaTK: Generates a wide range of fragmentomics features
- MethylDackel: Generates CpG, CHH, CHG bedgraph files
- Patr: Generates fragment-wise methylation pattern and summaries
- Stats: Consolidated statistics (Alignment, methylation, enrichment)
We have made this pipeline publicly available on our Strand Life Sciences Methylation Analysis Platform, which leverages AWS HealthOmics. Watch out for Part 2 of this series, which will provide a walkthrough of this portal for new users.


Let's Connect
Let's Connect

download the case study.