




















Blogs
Strand’s Automated Variant Verification System Cuts Down Efforts by 80%
In the past few decades, advancements in NGS have transformed the clinical genomics landscape. Cost-effective sequencing has given rise to an increasing number of SNVs and indels that contribute to diagnosis and treatment of inherited disorders and cancer.
A typical NGS workflow involves sample preparation, sequencing, QC, alignment, variant calling and annotation, a manual review, and, finally, interpretation and reporting. A crucial step in the process is variant calling as it determines the accuracy of the generated clinical report. In other words, variant calling tools compare aligned reads to the reference genome to identify SNPs and indels. Next, these variants are annotated, and their functional impact - known association with diseases and other relevant biological information - is recorded in a VCF file.
Given its high significance, expert bioinformaticians usually evaluate variants and filter out artifacts. Artifacts arise from genome assembly differences, homology sequencing errors, or pipeline parameters. While a careful and thorough review is necessary to avoid the risk of false reporting, this procedure, involving referring to various external tools and databases, is time-consuming. Not to forget, a manual review carries a level of subjective bias despite the scientists following specific guidelines.
To alleviate these concerns, at Strand, we have developed a modular system that drastically cuts short the time needed to verify a variant. This pluggable framework computationally evaluates variants and weeds out artifacts.
The step-by-step workflow leverages multiple tools - each responsible for a specific quality check. The quality checks cover a range of potential scenarios that give rise to artifacts. Here, we will dive into six of them -
- Supporting read quality check
- The check is passed if the following conditions are met at the variant location- there are at least 30 reads longer than 120 base pairs, each read has a mapping quality of 60, and at least 30% of these reads support the variant.
- Homopolymeric artifact check
- The homopolymer checker evaluates the genuineness of an indel variant by comparing the percentage of reads supporting the variant against a pre-computed error profile. If the supporting read percentage is below the threshold defined by the average plus two standard deviations, the variant is classified as an artifact.
- Homology artifact check
- The homology artifact checker determines if variant readings come from a homologous region using a pre-computed database. This database lists all unique bases that differentiate regions in the genome with over 90% similarity. The check is passed if the variant has at least five supporting read pairs that pass through at least 5 differentiating base loci containing reference bases.
- Assembly artifact check
- The assembly artifact checker selects a read containing the variant allele, aligns it to GRCh38 and T2T-CHM13 reference genomes, and evaluates these alignments to determine if the variant call results from differences between these genome builds
- Positional bias check
- The positional bias checker identifies whether the variant results from misalignment at the ends of the reads.
- Sequencing artifact check
- The sequencing artifact checker flags variants that are primarily supported by mates with mismatched bases at the variant location.
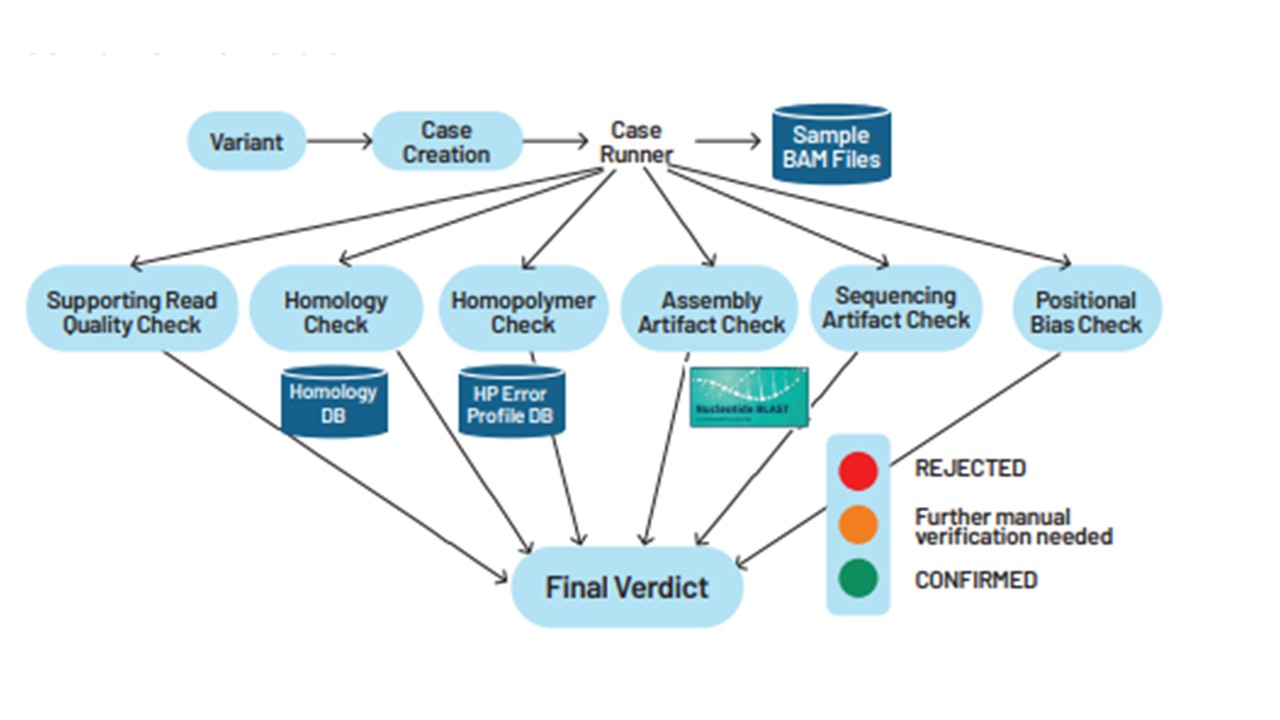
In summary, Strand’s automated variant verification system acts like a multipoint checklist, thus eliminating the possibility of artifacts leaking into clinical reports. This quality system not only saves time while reducing human error but also improves the accuracy of genetic analysis and clinical reports serving patients and researchers.

Precision Medicine
17 Nov 2025
How Strand is Connecting the Dots Across Individual Clinical Journeys

Precision Medicine
19 Feb 2024
Maximizing Data Power: The Role of Data Pooling in Pharma Research

Precision Medicine
09 Mar 2026
Strand’s Approach to Delivering Scalable, Clinical-Grade AI Infrastructure for Digital Histopathology
Let's Connect
Let's Connect

download the case study.